
Table of Contents
Gene networks inference
Objectives
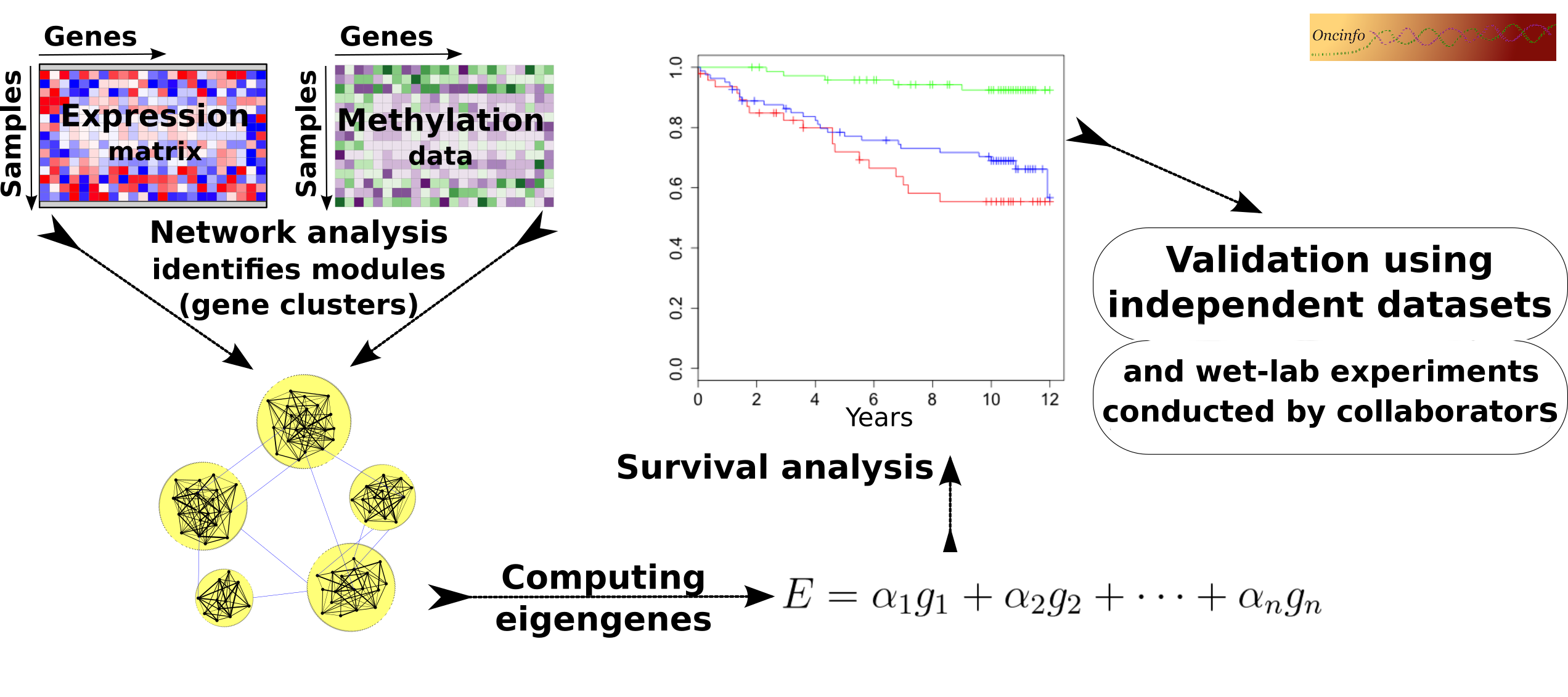
Biological processes in a cell often require intricate coordination between multiple genes and proteins. The goal of this project is to infer useful biological and clinical information from large networks of thousands of genes. We develop an integrative approach to analyze co-expression and DNA methylation patterns in a single model. The results will be useful in pinpointing the cause and mechanism of complex diseases such as cancer. Our findings can potentially open doors to new targets for novel treatment plans.
Collaborators
Dr. Aly Karsan from British Columbia Cancer Agency.
Papers
- A. Zainulabadeen et al., Underexpression of Specific Interferon Genes Is Associated with Poor Prognosis of Melanoma, PLoS One 2017, 12(1).
“Using our recently developed gene network model, we identified biological signatures that confidently predict the prognosis of melanoma. We showed that our predictive model assesses the risk more accurately than the traditional Clark staging method.” - Foroushani, Amir, et al. “Large-scale gene network analysis reveals the significance of extracellular matrix pathway and homeobox genes in acute myeloid leukemia: an introduction to the Pigengene package and its applications.” BMC medical genomics 10.1 (2017): 16.
- Agrahari, Rupesh, et al. “Applications of Bayesian network models in predicting types of hematological malignancies.” Scientific Reports 8.1 (2018): 6951.
Software
Pigengene
This R package provides an efficient way to perform network analysis and to infer biological signatures from gene expression profiles. The signatures are independent from the underlying platform, e.g., it can infer the signatures using data from microarray and evaluate them in an independent RNA Seq dataset. It is approved by, and publicly available from, Bioconductor.
iNETgrate
This R package is useful to integrate DNA methylation and gene expression data into a single network (code). This approach leads to identification of more robust gene modules compared to conventional coexpression networks. The package will be publicly available after review and approval by Bioconductor. A checklist for package completion tasks.
Data
- mRNA expression and Mutation data from Novartis.
Broad-Novartis Cancer Cell Line Encyclopedia (CCLE, Barretina et al.).About 500 cell lines from different human cancers. The goal here is to predict drug responses in particular in synergistic settings. Specifically, BROWS > DATA >
a. mRNA expression > gene-centric RMA-normalized mRNA expression data > the gctx file. (How to read a gctx file?)
b. Pharmacological profiling Drug data > Pharmacologic profiles for 24 anticancer drugs across 504 CCLE lines.
c. Clustering and gene ontology analysis done by Dr. Chris Gaiteri. - RNA-seq from about a hundred AML and MDS cases are available from Karsan lab. The goal here is to identify the general underling mechanisms of the disease, and to compare them with the relapse factors. More specific questions are a) What pathways are different in AML than MDS? b) Are there pathways which can define AML subtypes, which are expected to exist due to differences in prognosis? c) What are the molecular mechanisms of transformation of some MDS cases to AML?
- Genentech data set published in 2014 (Klinj et al.), RNA-seq for 675 cell lines including 15 AMLs, and response to 5 drugs.
- Sanger data set published in 2012 (Garnet et al.), similar to CCLE data with 639 cell lines and 130 compounds.
- GlaxoSmithKline data set published in 2010 (Greshock et al.), similar to CCLE data with 311 cell lines and 19 compounds.
- Nucleic Acids Research online Molecular Biology Database Collection.
lists 1512 miscellaneous online databases. - AstraZeneca's crowd sourcing initiative as part of the DREAM Challenge. ~10,000 tested combinations measuring the ability of drugs to destroy cancer cell lines, and the corresponding genomic information.
- Breast Cancer datasets: We will examine the generalizability of the method that we developed for haematological malignancies (AML/MDS) by examining its performance on several breast cancer datasets: 209 ER samples from Wang et al's dataset (GEO). (Paper), 201 ER samples from Miller et al's dataset (GEO), as well as expression data from METABRIC study ( ~2000 samples, hosted by EGA) (paper).
- Microarray expression profiles of 1005 colorectal cancer patients from 13 independent cohorts (paper).
- Gene expression data of fish exposed to light (Walter Lab).
- 16 pairs of tumor-normal samples from fish with melanoma (Walter Lab).
- 499 prostate adenocarcinoma (TCGA, Provisional) samples. Low risk cases are “Disease Free” for at least 5 years and the “Recurred” ones are high risk. The relevant clinical data are shown in “Disease Free (Months)” and “Disease Free Status” columns in cBioPortal, respectively.
- 470 skin cutaneous melanoma samples from TCGA. The clinical data for survival analysis are shown in “Disease Free (Months)”, “Disease Free Status”, and “Days to Last Followup” columns.
- German AMLCG 1999 provides microarray data of 562 AML samples.
- Papaemmanuil, Elli, et al. “Genomic classification and prognosis in acute myeloid leukemia.” NEJM 374.23 (2016): 2209-2221.
The mutations of 111 genes in over 1,500 AML cases are reported. The authors used this information to classify cases into groups and showed these groups have different prognosis. I.,e., concordance (probability estimates) improves from 64% using only the European LeukemiaNet criteria to 71%. Using the alternative allele frequency, they estimated the time of occurrence for the driver mutations. The data are available through the links in the corresponding Nature paper pdf]. Information on downloading these data is contained in the readme file found in genetwork:~/proj/genetwork/data/AML/gerstung/readme.txt. In particular, we have access to EGAS00001000275 through EGA Archives. See Habil's note on 2017/09/05 for more detail. Any member of Oncinfo Lab who touches (analyzes or views) these data from Sanger Institute must read and abide to the agreement. - RNA, DNA methylation, whole genome, etc. data of 960 (pediatric?) AML cases are available from TARGET AML study.
- AML-NK gene expression data (RNA-Seq) from three datasets (TCGA, Leucegene, and PMP/BCCA). Full description.
- List of available AML datasets with DNA methylation or gene expression data.
- Genomic Data Commons (GDC), which contains TCGA data and more.
- ARCHS4, which was developed at the Icahn School of Medicine at Mount Sinai, and provides tools to download and analyze RNA-Seq data including single-cell gene expression.
- The BEAT ALM dataset of ~300 cases including gene expression, survival, ELN17, etc.
- Leukemia Protein Atlas: Expression of hundreds of proteins were measured in bone marrow and PB samples of ~200 AML cases. A good publicly available resource to validate findings based on gene expression assays.
- ~40K single cell RNA-Seq data from 40 bone marrow aspirates, including 16 AML patients and
5 healthy donors.
{kind=link}
Related work
- Zhang, B. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell153, 707–720 (2013). [pdf ]
Methodology is based on [2] plus they train Bayesian networks to infer causal structure based on RNA-seq. MCMC was used to optimize BIC score, and
1/3 of 1000 network models were averaged.This webinar is a high level description of the methodology. - Integrating Genetic and Network Analysis to Characterize Genes Related to Mouse Weight (Identified modules in networks, “A pair of genes is said to have high topological overlap if they are both strongly connected to the same group of genes.”, Software)
- Gene correlation network analysis [Wikipedia page]
- Friedman, Nir, et al. “Using Bayesian networks to analyze expression data.” Journal of computational biology 7.3-4 (2000): 601-620.[pdf ]
A relatively old but highly cited, (~2700) original paper. - Ruan, Jianhua, Angela K. Dean, and Weixiong Zhang. “A general co-expression network-based approach to gene expression analysis: comparison and applications.” BMC systems biology4.1 (2010): 8. (Dr. Jianhua Ruan from San Antonino)
- Nagrecha, Saurabh, Pawan J. Lingras, and Nitesh V. Chawla. “Comparison of gene co-expression networks and bayesian networks.” Intelligent Information and Database Systems . Springer Berlin Heidelberg, 2013. 507-516.
Simple description and some relatively old literature review.
“Bayesian networks emerge as a more informative tool to determine the causal structure.” - Systems Biology: The inference of networks from high dimensional genomics data, lecture by Yeung 2011 [ppt ] (a good introduction to application of Bayesian networks in co-expression network)
- Hong, Shengjun, et al. “Canonical correlation analysis for RNA-seq co-expression networks.” Nucleic acids research41.8 (2013): e95-e95. (improved co-expression analysis for RNA-seq)
- Li, Bingshan, et al. “Transcriptome Analysis of Psoriasis in a Large Case–Control Sample: RNA-Seq Provides Insights into Disease Mechanisms.” Journal of Investigative Dermatology (2014). (used co-expression network analysis on RNA-seq from 42 samples to study gene regulatory circuits in psoriasis)
- Analysis of RNA‐Seq Data, an introduction by Wong, 2010.
- Ellis, Byron, and Wing Hung Wong. “Learning causal Bayesian network structures from experimental data.” Journal of the American Statistical Association 103.482 (2008): 778-789.
- Barretina et al. “The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity.” Nature 483.7391 (2012): 603-607 [pdf ].
It provides CCLE, a valuable dataset produced by Novartis; mRNA expression data, responses of 24 compounds, and targeted sequencing on ~500 cell lines. - Friedman, Nir, et al. “Using Bayesian networks to analyze expression data.”Journal of computational biology7.3-4 (2000): 601-620.
Has a good introduction to Bayesian networks and learning causal patterns for beginners. - Al-Lazikani, Bissan, Udai Banerji, and Paul Workman. “Combinatorial drug therapy for cancer in the post-genomic era.” Nature biotechnology30.7 (2012): 679-692.
“Combinatorial targeted therapy”, a good survey including computational methods, successful stories like “identification of synergies between MET and EGFR inhibitors…” - Molinelli, Evan J., et al. “Perturbation Biology: inferring signaling networks in cellular systems.” PLoS computational biology9.12 (2013): e1003290.
“…inferring network models that predict the response of cells to perturbations which may be useful in the design of combinatorial therapy against cancer. Claims differential equations work better than Bayesian networks (Biolearn). - Marbach, Daniel, et al. “Revealing strengths and weaknesses of methods for gene network inference.” PNAS 107.14 (2010): 6286-6291.
Survey on DREAM results, includes Bayesian network methods. - Molinelli, Evan J., et al. “Perturbation Biology: inferring signaling networks in cellular systems.” PLoS computational biology 9.12 (2013): e1003290.
Belief Propagation technique developed in Sloan Kattering, claimed to be better than BN. - Zhao, J., X. S. Zhang, and S. Zhang. “Predicting cooperative drug effects through the quantitative cellular profiling of response to individual drugs.” CPT: pharmacometrics & systems pharmacology 3.2 (2014): e102.
“By exploring the differential expression profiles, our correlation-based strategy can reveal the synergistic effects of drug combinations.” A recent paper which used a simple method and includes a good literature review. A good reference to datasets similar to CCLE. - Du, W., and O. Elemento. “Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies.” Oncogene (2014).
A recent review paper explaining the motivation and need for computational methods. - Han, Bing, and Xue-wen Chen. “bNEAT: a Bayesian network method for detecting epistatic interactions in genome-wide association studies.” BMC genomics 12.Suppl 2 (2011): S9.
Potential collaboration with Chen. Argues that Branch-and-Bound is better than MCMC. Good introduction to BN structure learning. - Bayesian Network Learning and Applications in Bioinformatics (2012),
Xiaotong Lin's PhD thesis with a good introduction to the basic concepts and challenges. - Bansal, Mukesh, et al. “How to infer gene networks from expression profiles.”Molecular systems biology 3.1 (2007).
A relatively old survey which supports Banjo. - Mourad, Raphaël, and Christine Sinoquet, eds. Probabilistic Graphical Models for Genetics, Genomics and Postgenomics. Oxford University Press, 2014.
An excellent recent, relevant book ( e.g. pages 24,123,154,223).P155: For gene networks, maximum number of parents is commonly set to 3. - de la Fuente, Alberto. “Gene Network Inference.”, Springer, 2013 [pdf ].
Figure 1 explains why a network-based approach can be superior to black box machine learning techniques. Literature review on advantages of system approaches over studying single genes. Limited the maximum number of parents per gene to 5. Zhu et al. explains RIMBANet developed by himself. “Bayesian networks offer the best performance.” - Bayesian Networks with R (bnlearn) and Hadoop, 2014, a good talk by
Ofer Mendelevitch with introduction to BNs. Discusses large networks too. - Network Analysis Workshop, Systems Biology Analysis Methods for Genomic, 2013, UCLA. Talk in Bayesian networks by Jun Zhu.
- Vignes, Matthieu, et al. “Gene regulatory network reconstruction using bayesian networks, the dantzig selector, the lasso and their meta-analysis.” PloS one 6.12 (2011): e29165.
A meta-analysis to combine these inference methods by computing a consensus ranking scheme, ranked 1st among 16 in a DREAM challenge, but its superiority was not confirmed in Allouche 2014. Used greedy hill-climbing of Banjo. - Schadt, Eric E., et al. “An integrative genomics approach to infer causal associations between gene expression and disease.” Nature genetics 37.7 (2005): 710-717.
The original Schadt paper introducing the idea of using genomic data to infer causality in BNs of expression (LCMS). - Jiang, Xia, et al. “Learning genetic epistasis using Bayesian network scoring criteria.” BMC bioinformatics 12.1 (2011): 89.
On simulated data, Bayesian scoring (BDeu) outperforms minimum description scores such as AIC. - Hartemink, Alexander John. Principled computational methods for the validation and discovery of genetic regulatory networks. Diss. MIT, 2001.
Hartemink's (author of Banjo) thesis, well explains BDe score and Dirichlet priors. - Yu, Jing, et al. “Advances to Bayesian network inference for generating causal networks from observational biological data.” Bioinformatics 20.18 (2004): 3594-3603.
From Hartemink group, ~500 citations. DBN. - Yu, Jing, et al. “Using Bayesian network inference algorithms to recover molecular genetic regulatory networks.” 3rd International Conference on Systems Biology. 2002.
From Hartemink group, DBN. Very good explanation and comparison of different scores and search strategies. “With large amounts of data, the BIC is a good approximation to the full posterior (BDe) score and is faster to compute; however, it is known to over-penalize with small amounts of data.” “The BDe score works better than the BIC score in recovering genetic regulatory pathways.” [Compared to simulated annealing and genetic algorithm,] greedy search is better as it can find the top graph in the least amount of time” (note that their network is small). “ 3-category discretization was optimal. - Zhu, Jun, et al. “Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations.” PLoS computational biology 3.4 (2007): e69.
The methodology used to learn BNs in Zhang et al. 2013 AD paper. - Zhu, J., et al. “An integrative genomics approach to the reconstruction of gene networks in segregating populations.” Cytogenetic and genome research 105.2-4 (2004): 363-374 [pdf ].
The methodology used to incorporate genetic data to better learn BNs (e.g. useful in Zhang et al. 2013 AD paper). Edges which appeared in more than 30% of 1000 graphs should be used to make the consensus graph. - Steidl, Ulrich G., and Constantine S. Mitsiades. “Therapeutic and diagnostic target gene in acute myeloid leukemia.” U.S. Patent Application 14/113,405.
Useful for sanity check to see if the genes we identify are known to be important. - Kommadath, Arun, et al. “Gene co-expression network analysis identifies porcine genes associated with variation in Salmonella shedding.” BMC genomics15.1 (2014): 452.
A recent study that used WGCNA on RNA-seq. - Oldham, Michael C., et al. “Functional organization of the transcriptome in human brain.” Nature neuroscience11.11 (2008): 1271-1282.
WGCNA authors used it to provide biological insight. Validated co-expression with experimentally validated interacting human protein pairs from EBI slides]. - Iancu, Ovidiu D., et al. “Utilizing RNA-Seq data for de novo coexpression network inference.” Bioinformatics28.12 (2012): 1592-1597.
“The first RNA-Seq data de novo network inference…. Retained the probes with above median connectivity, resulting in 3618 common probes.” - Klijn, Christiaan, et al. “A comprehensive transcriptional portrait of human cancer cell lines.” Nature biotechnology (2014).
RNA-seq data for 675 cell lines, also a good and recent source for references and similar data such as CCLE and Sanger 2012 data set. “… clustering of RNA-seq–derived gene expression showed that …the lymphoid cells formed a notably distinct cluster” So there should be potential for interpreting by modules. - Garnett, Mathew J., et al. “Systematic identification of genomic markers of drug sensitivity in cancer cells.” Nature483.7391 (2012): 570-575.
Sanger data set similar to CCLE data with 639 cell lines and 130 compounds. - Haibe-Kains, Benjamin, et al. “Inconsistency in large pharmacogenomic studies.” Nature (2013).
“The measured drug response data are highly discordant” between CCLE and Sanger data! - Soneson, Charlotte, and Mauro Delorenzi. “A comparison of methods for differential expression analysis of RNA-seq data.” BMC bioinformatics14.1 (2013): 91.
- Friedman, Nir, and Daphne Koller. “Being Bayesian about network structure. A Bayesian approach to structure discovery in Bayesian networks.” Machine learning 50.1-2 (2003): 95-125.
~ 600 citations. Tries to address the challenge of learning a large network (thousands of genes) with few (hundreds of) samples using a Bayesian approach. Also, learning a total ordering is suggested. Implemented in catNet. - Subramanian, Aravind, et al. “Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.” PNAS 102.43 (2005): 15545-15550.
Over 7K citations. Microarray on mRNA. Criticizes DE analysis. Gene sets are given based on prior knowledge and scored based on association with the phenotype. - Choi, YounJeong, and Christina Kendziorski. “Statistical methods for gene set co-expression analysis.” Bioinformatics 25.21 (2009): 2780-2786.
Criticizes approaches that are based on gene modules. Identifies changes in gene correlations. Uses gene sets in KEGG and GO. Comparison and introduction to well-known gene set enrichment methods till 2009. - Ideker, Trey, et al. “Discovering regulatory and signalling circuits in molecular interaction networks.”Bioinformatics18.suppl 1 (2002): S233-S240.
“Screening a molecular interaction (protein-protein) network to identify active (altered) subnetworks, i.e., connected regions of the network that show significant changes in expression over particular subsets of conditions.” Input: expressions pathways data base, output: altered subnetworks of each pathway. - Gene Set Enrichment Analysis (GSEA) is a computational method by Broad that determines whether an a priori defined set of genes (e.g. MSigDB) shows statistically significant, concordant differences between two biological states (e.g. phenotypes).
Input: expressions pathway data base, output: p-values for each “pathway”. - Isci, Senol, et al. “Bayesian network prior: network analysis of biological data using external knowledge.”Bioinformatics30.6 (2014): 860-867.
Gene interaction networks are learned from experimental data using Bayesian networks (BNs) while incorporating external knowledge, such as KEGG and NCI, as Bayesian Network Prior (BNP). Includes links to several useful data sets. Build on their former approach for improving pathways by BNs. Used fold change discretized by kmeans as the value of each random variable (gene).Input: expressions, output: “pathways”. - Nagarajan, Radhakrishnan, et al. “Functional relationships between genes associated with differentiation potential of aged myogenic progenitors.” Frontiers in Physiology 1 (2010).
Proposed a method to identify the threshold on the strength of edges in a consensus BN based on background noise. - Niculescu, Radu Stefan, Tom M. Mitchell, and R. Bharat Rao. “Bayesian network learning with parameter constraints.” The Journal of Machine Learning Research 7 (2006): 1357-1383.
“Automatically discovering clusters of voxels that can be more accurately learned with shared parameters” - Gillis, Jesse, and Paul Pavlidis. ““Guilt by association” is the exception rather than the rule in gene networks.” PLoS computational biology 8.3 (2012): e1002444.
Discussed the difficulties of computational network analysis. “…Functional information within gene networks is typically concentrated in only a very few interactions whose properties cannot be reliably related to the rest of the network”. - Koski, Timo JT, and John Noble. “A review of bayesian networks and structure learning.” Mathematica Applicanda 40.1 (2012): 51-103.
A review from mathematical view point including applications of algebraic geometry to Bayesian networks! - Barabási, Albert-László, Natali Gulbahce, and Joseph Loscalzo. “Network medicine: a network-based approach to human disease. (pdf )” //Nature Reviews Genetics// 12.1 (2011): 56-68.
- Kustra, Rafal, and Adam Zagdanski. “Incorporating gene ontology in clustering gene expression data.” Computer-Based Medical Systems, 2006. CBMS 2006. 19th IEEE International Symposium on. IEEE, 2006.
- Dotan-Cohen, Dikla, Simon Kasif, and Avraham A. Melkman. “Seeing the forest for the trees: using the gene ontology to restructure hierarchical clustering.”//Bioinformatics// 25.14 (2009): 1789-1795.
Semi-supervised clustering. - see the graphic under section two here.Kang, Bo-Yeong, Song Ko, and Dae-Won Kim. “SICAGO: Semi-supervised cluster analysis using semantic distance between gene pairs in Gene Ontology.” //Bioinformatics// 26.10 (2010): 1384-1385.
- Mostafavi, Sara, et al. “GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function.” //Genome Biol// 9.Suppl 1 (2008): S4. Website
Predicting gene function using prior biological knowledge and expression. - Frost, Hildreth R., Zhigang Li, and Jason H. Moore. “Spectral gene set enrichment (SGSE).” BMC bioinformatics16.1 (2015): 70.
Enrichment analysis on PCA. - Soneson, Charlotte, and Mauro Delorenzi. “A comparison of methods for differential expression analysis of RNA-seq data.” BMC bioinformatics14.1 (2013): 91.
Compared 11 methods including DESeq and edgR. Before remove (RUV), which estimated expression in compassion to controls. - Wagner, Günter P., Koryu Kin, and Vincent J. Lynch. “Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples.” Theory in Biosciences 131.4 (2012): 281-285.
Use TPM instead forum, conversion]. - Horvath, Steve. Weighted Network Analysis: Applications in Genomics and Systems Biology. Springer Science & Business Media, 2011.
An overview on network analysis by the author of WGCNA. - Conway O’Brien, Emma, Steven Prideaux, and Timothy Chevassut. “The epigenetic landscape of acute myeloid leukemia.” Advances in hematology 2014 (2014).
Review paper. Talks about H3K27me and other histone modifications in AML. Also see references 73 (knocked down ASXL1 in primary hematopoietic cells and measured H3K27me3), 6 (review), 8 (review), and 76 (studied the role of EZH2). - Yamazaki, Jumpei, et al. “The epigenome of AML stem and progenitor cells.” Epigenetics 8.1 (2013): 92-104.
“Found no major differences in DNA methylation” but “thousands of genes that change H3K4me3 or H3K27me3” - Abdel-Wahab, Omar, et al. “ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression.” Cancer cell 22.2 (2012): 180-193.
Includes Chip-seq analysis of H3K27me3 on AML. - Halsey, Lewis G., et al. “The fickle P value generates irreproducible results.” Nature methods 12.3 (2015): 179-185.
A reference for justifying the need for network analysis. - Wang, Bo, et al. “Similarity network fusion for aggregating data types on a genomic scale.”Nature methods11.3 (2014): 333-337.
Combined mRNA expression, DNA methylation and microRNA (miRNA) expression data. Built a patient network for each data type using Euclidean distance with a Gaussian kernel. Combined the networks using message-passing. Applied on METABRIC and compared with a couple of other methods. - Reid, Stephen, and Robert Tibshirani. “Sparse regression and marginal testing using cluster prototypes.” Biostatistics (2015).
Read this recent paper to follow the literature after LASSO. - Torkamani, Ali, and Nicholas J. Schork. “Identification of rare cancer driver mutations by network reconstruction.” Genome research 19.9 (2009): 1570-1578.
“Although there are many algorithms and strategies for reconstructing gene coexpression networks, including those that exploit a simple pairwise correlation matrix of gene-expression levels and clustering algorithms, the mutual information approach in the ARACNE algorithms has been shown to provide superior results in related contexts.” - Wang, Jinlian, et al. “Pathway and network approaches for identification of cancer signature markers from omics data.” J Cancer6.1 (2015): 54-65.
Surveyed integrating methods too. - Hill, Steven M., et al. “Inferring causal molecular networks: empirical assessment through a community-based effort.” Nature methods (2016).
A DREAM challenge. Compared 2000 networks in 32 biological contexts. - Manning, Cerys Sian. Heterogeneity in melanoma and the microenvironment. Diss. UCL (University College London), 2013.
A PhD thesis with a good introduction to melanoma. - Shannan, Batool, et al. “Heterogeneity in Melanoma.” Melanoma. Springer International Publishing, 2016. 1-15 pdf].
A chapter of a comprehensive recent book on melanoma. - Jiao, Yinming, Martin Widschwendter, and Andrew E. Teschendorff. “A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control.” Bioinformatics 30.16 (2014): 2360-2366.
FEM paper. - Guillamot, Maria, Luisa Cimmino, and Iannis Aifantis. “The impact of DNA methylation in hematopoietic malignancies.” Trends in cancer 2.2 (2016): 70-83.
Reviews and references DNA methylation studies and datasets on AML. E.g., Figueroa et al. used DNA methylation for classification of 344 AML cases. Akalin et al. related DNA methylation patterns with mutations in 5 AML cases. “The methylation status of specific genes can predict the future survival of AML patients, suggesting that DNA methylation is a biomarker for clinical outcome” see e.g., Figueroa et al, Jiang 2009 (studied MDS to AML progression in 184 cases), and Bullinger 2010 (analyzed 92 genomic regions in 182 patients). - John Quackenbush's talk entitled: “Using Networks to Understand the Genotype-Phenotype Connection”.
- Saelens, Wouter, Robrecht Cannoodt, and Yvan Saeys. “A comprehensive evaluation of module detection methods for gene expression data.” Nature communications 9.1 (2018): 1090.
“Graph-based, representative-based, and hierarchical clustering all performed equally well, with the clustering method FLAME (Fuzzy clustering by Local Approximation of Memberships), one of the only clustering methods able to detect overlap, slightly outperforming other clustering methods” including WGCNA. Regularity networks that had been inferred using other data, e.g., “binding motifs in active enhancers”, were used as gold standard. - Choobdar, Sarvenaz, et al. “Assessment of network module identification across complex diseases.” Nature Methods 16.9 (2019): 843-852.
“The popular weighted gene co-expression network analysis (WGCNA) method7 did not perform competitively.”
Related software
- Weighted Gene Co-expression Network Analysis (WGCNA) developed at UCLA. The page has links to some good introductory workshops.
- birta (Bayesian Inference of Regulation of Transcriptional Activity) uses the regulatory networks of TFs and miRNAs together with mRNA and miRNA expression data to predict switches in regulatory activity between two conditions. A Bayesian network is used to model the regulatory structure.
- GRENITS, Regulatory Network Inference Using Time Series, is based on Dynamic Bayesian Networks.
- BANJO, a Java software for structure learning of static and dynamic Bayesian networks developed at Duke University paper (DBN)]. No inference, BDe score, simulated annealing and greedy searches, handles thousands of variables, used for gene network inference. “In our experience the simulated annealer method [with RandomLocal] seems to produce the best results.” Computes consensus graph, discretization, Forces or disallows edges by “network structure properties” which is useful for merging networks.
- Biolearn, applies probabilistic graphical models to biological data. Not maintained anymore.
- deal: an R package for Learning Bayesian Networks. Greedy search.
- Software Packages for Graphical Models (2007), Murphy's list. See our refined comparison of Bayesian network learners.
- gr, gRaphical Models in R
- RIMBANet, reconstructs integrative molecular Bayesian networks, maybe used to study Alzheimer’s Disease in the Cell paper.
- bnlearn, an R package for learning BNs which claims to learn a ~1000-nodes network (introductory slides). A descriptive comparison with other R packages. No MCMC. See Radhakrishnan et al. above.
- Cytoscape, visualizes molecular interaction networks and biological pathways.
- See the table for comparison of methods for learning BNs related to our gene network project.