
Table of Contents
Hepatocellular carcinoma
Objectives
Hepatocellular carcinoma (HCC) is a the most common type of liver cancer and the third leading cause of cancer deaths in the world. Cirrhosis of the liver is a major risk and contributing factor for HCC. The goal of project one is to identify the genes that are associated with regressing tumors (regardless of type of treatment) vs. those that are growing from the C3HeB/FeJ mouse model. Normal liver is used as a control. The results will be useful in identifying new therapeutic targets and potential drug combinations which could lead to more efficient treatments. Project two is related to knockdown of a specific protein that results in HCC in a different mouse model. The goal in the second project is to identify the genes and pathways that correlate with this protein.
Sources of Human HCC RNA-seq Data
- GSE25599 10 match-paired HBV-related Chinese HCC and non-cancerous adjacent tissues. Identified 1,378 significantly DE genes
- GSE33294 Chinese HBV-related hepatocellular carcinoma, paired tumor and non-cancerous adjacent tissues from 3 patients.
- GSE59259 Alcohol-related HCC 8 paired samples of liver and HCC
- SRA074279 9 Chinese patients: paired HCC and adjacent non-cancerous tissues; Publication
- GSE55759 Paired HCC and non-cancerous adjacent tissue; median of 7019 DE genes per set, 93 DE genes shared by 6/8 patients
| Sex/Age | Viral Infection | Tumor Differentiation | No. of Tumors | Vascular Invasion | TNM* Stage |
| M/62 | HBV(-)HCV(-) | Well | 1 | No | II |
| F/29 | HBV(+)HCV(-) | Well | 1 | No | II |
| M/56 | HBV(+)HCV(-) | Moderately | No | II | |
| M/55 | HBV(+)HCV(-) | Moderately | 1 | No | II |
| F/39 | HBV(+)HCV(-) | Moderately | 1 | No | II |
| M/44 | HBV(+)HCV(-) | Moderately | 1 | Yes | III |
| M/47 | HBV(+)HCV(-) | Moderately | 1 | No | II |
| M/48 | HBV(+)HCV(-) | Poorly | 1 | Yes | III |
Collaborators
Drs. Christi Walter and Lily Dong from the Department of Structural and Cellular Biology at UT Health Science Center in San Antonio.
Analysis
Project 1:
- Uploading RNAseq data to Maverick: Jessica's lab notebook, 2015/12/15.
- Mapping reads and de analysis: Habil's lab notebook, 2015/12/16.
- Mapping gene names: Amir's lab notebook, 1/10/2016.
- Computing eigengen: Habil's lab notebook, 2016/01/27
- Comparing de genes with human databases: Jessica's lab notebook, 2016/2/4.
- The comparison of Will's DE analysis and ours: Jessica's lab notebook, 2016/2/4.
- PCA: Jessica's lab notebook, 2016/5/31.
- Habil inferred the eigengene in TCGA data on 2019/05/14.
Publication:
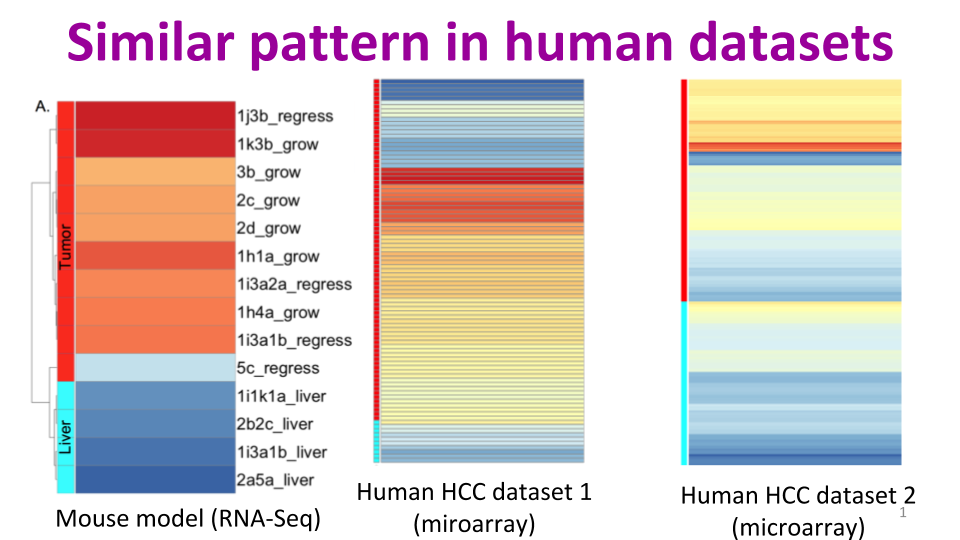
Jessica Zavadil, Maryanne Herzig, Kim Hildreth, Amir Foroushani, William Boswell, Ronald Walter, Robert Reddick, Hugh White, Habil Zare. “C3HeB/FeJ Mice mimic many aspects of gene expression and pathobiological features of human hepatocellular carcinoma.” Molecular carcinogenesis 58.3 (2019): 309-320.
Project 2:
Project 3:
Protemoe and the APEX1 interactome: Data-independent analysis Mass spectrometry (DIA-MS) done on 3 biological replicates of 2 HCC cell lines (SNU398 and Huh7) and an immortalized hepatocyte line (THLE2). Also, we have 1 biological replicate of a primary hepatocyte organoid derived from a patient (UTHSS-28T).
Our goal is to understand if the APE1 interactome is 1) different in HCC cell lines vs non tumor cells, 2) different between two HCC cell lines with overexpressed APEX1 (SNU398 vs Huh7), 3) and how it compares to that described in Ayyildiz 2020. Christi sent these data to Habil on 2020-12-02 in an email entitled: “M2021-026 Scaffold DIA and Excel files”.
Related work
- Hoenerhoff, Mark J., et al. “Global Gene Profiling of Spontaneous Hepatocellular Carcinoma in B6C3F1 Mice Similarities in the Molecular Landscape with Human Liver Cancer.” Toxicologic pathology39.4 (2011): 678-699.
Microarray analysis of tumors from B6C3F1 mice (first generation of C57B/L6J and C3HeB/FeJ strains) - Keane, Thomas M., et al. “Mouse genomic variation and its effect on phenotypes and gene regulation.” Nature 477.7364 (2011): 289-294.
Compared the standard reference genomes of mouse (C57BL/6J) with other strains. - Munger, Steven C., et al. “RNA-Seq alignment to individualized genomes improves transcript abundance estimates in multiparent populations.”Genetics 198.1 (2014): 59-73.
Proposed a method for strain-specific alignment and compared with mapping RNAseq data from a strain to the reference genome. Observed >10% change in expression in about 2,000 genes. - Huang, Shunping, et al. “Transforming genomes using MOD files with applications.” Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics. ACM, 2013.
Figure 4 shows that if we map to reference genome, we may loose not more than 7% of reads. - Hart, Steven N., et al. “Calculating sample size estimates for RNA sequencing data.” Journal of Computational Biology20.12 (2013): 970-978.Wu, Hao, Chi Wang, and Zhijin Wu. “PROPER: comprehensive power evaluation for differential expression using RNA-seq.” Bioinformatics31.2 (2015): 233-241.From Fig2 and Fig3 of Huang et al. paper, and Fig5 of Hart et al., it seems that at least 5-7 samples are needed for each condition.
- Ching, Travers, Sijia Huang, and Lana X. Garmire. “Power analysis and sample size estimation for RNA-Seq differential expression.” rna20.11 (2014): 1684-1696.
- Subramaniam, Somasundaram, Robin K. Kelley, and Alan P. Venook. “A review of hepatocellular carcinoma (HCC) staging systems.” Chinese clinical oncology 2.4 (2013).
- Alexandrov, Ludmil B., et al. “The repertoire of mutational signatures in human cancer.” Nature 578.7793 (2020): 94-101.
Analyzed WGS and WXS data of thousands of tumors available from TCGA and PCAWG consortia. - Dr. Sukeshi Arora's slides presented in the HCC meeting on 2020-04-18, which summarizes statistics on the prognosis, the current clinical practice, and response to different treatments.
Related software
- TopHat, useful for aligning RNAseq data to a genome.
- StringTie, reconstructs transcriptom from RNAseq data (2015).
- Homer's quick tutorial on mapping NGS data using several tools including bowtie2, bwa, TopHAt, etc. with command line examples.
- Lefebvre's quick tutorial on RNA-Seq data analysis.
- Schiffthaler's ~1 hour video on RNA Seq data preprocessing including FastQC, sortmerna to exclude rRNA, trimmomatic to trim the adaptors and low quality bps, STAR to map reads to the genome, samtools to index the bam file, IGV to visualize the reads on the genome, and HTSeq to count the number of reads mapped to each gene (coverage). These are all steps we need to do before differential analysis using, say DESeq2. This is a textual version explaining the same steps.